
2 一般而言,注射用粉末复溶后,溶液体积会增加。增加的体积应符合中国药典通则中关于注射液适当增加的灌装量,如不符合,可通过可转移体积等研究说明其合理性,确保每瓶产品的实际给药剂量均不低于标示量。
应采用多批商业化规模生产样品进行下述研究:
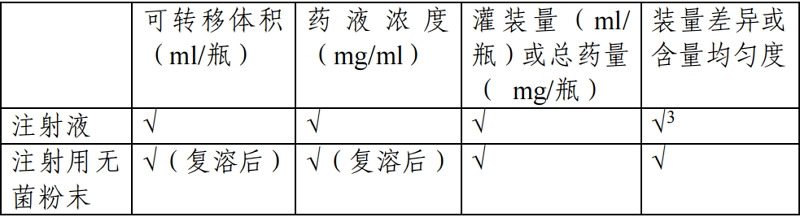
申请人应提供仿制药与参比制剂可转移剂量的对比研究资料,仿制药的可转移剂量应满足说明书中标示的给药剂量。
其他类型的注射剂可参考本指导原则的研究思路开展研究。
四、申报资料要求
申请人应在申报资料中提供过量灌装的相关研究资料。过量灌装量应在 CTD 通用技术文档“3.2.P.1 剂型及产品组成”部分进行描述;相关研究验证资料(如,可转移体积测试、黏度研究、灌装量范围等)和过程控制应在 CTD 通用技术
文档“3.2.P.2 产品开发、3.2.P.3 生产”部分进行描述。
五、说明书和标签
如适用,建议参照参比制剂说明书,在【用法用量】项下标注复溶后药液浓度。为避免临床用药困惑,过量灌装量通常不需要在标签和说明书中特别说明。
3 可根据中国药典要求执行。
六、名词解释
可转移体积:系指根据中国药典规定的【装量】检查方法或其他适宜且经过验证的方法,液体药物或固体药物复溶后可以从原容器中转移的体积。
可转移剂量:系指根据可转移体积和药物浓度(注射液)/复溶后药物浓度(注射用无菌粉末)计算得到的活性成分的量。
灌装量:系指西林瓶或安瓿瓶中注射液的液体总量(如,ml),包括标示装量和增加的体积。
总药量:系指西林瓶中的注射用无菌粉末中活性成分的总量(如,mg),包括标示装量和增加的药量。
标示量: 系指说明书和标签中标示的活性成分的量。
七、参考文献
1.中国药典 2020 年版四部通则 0102
2.国家药品监督管理局. 化学药品注射剂仿制药质量和疗效一致性评价技术要求. 2020
3. FDA. Guidance for Industry:Allowable Excess Volume and Labeled Vial Fill Size in Injectable Drug and Biological Products.2015
4.FDA. Allowable Excess Volume/Content in Injectable Drug and Biological Products(MAPP 5019.1 Rev 1).2022
5.ICH Steering Committee, Harmonised Tripartite Guideline Q8(R2): Pharmaceutical Development. August, 2009
6. USP<1151> Pharmaceutical dosage forms
7. USP< 697> Container content for injections
