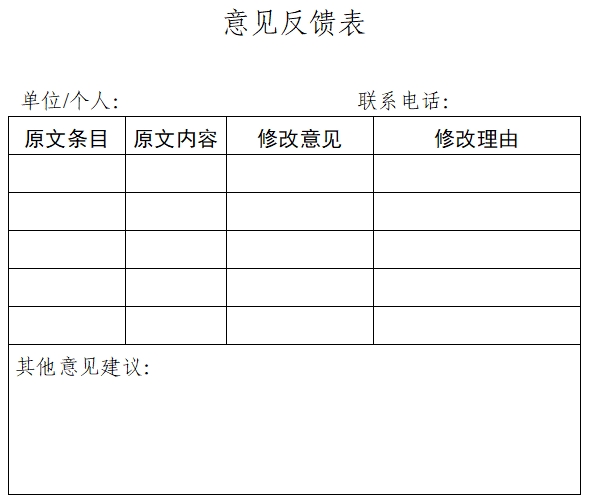
4.受托生产企业同剂型及生产线近三年未发生产品抽检不合格、生产检查结论不符合药品GMP要求等违法违规情形说明;无菌药品委托生产的,持有人和受托生产企业双方同剂型药品商业化生产经验情况说明;
5.受托生产企业自评估报告:包括设施设备与申请品种工艺匹配情况、拟受托品种同类型制剂药品实际生产经验情况、共线生产风险评估及相关持有人审批情况、产能情况(包括共线品种数量、各品种近两年生产批次、批量情况,拟受托生产后生产线产能匹配情况)、对持有人的质量管理系统进行评估审核情况(包括委托方质量管理体系运行情况、技术转移管理能力、受托生产品种的风险因素、分析共线生产的可行性等)、不良信用情况等;
6.委托双方的委托合同和质量协议复印件(涉及商业机密的可不显示);
7.双方关键人员履历;
8.持有人对受托生产企业的评估确认报告:包括对其机构与人员、生产设备、生产经验、生产线产能、检验能力(如需要)和共线生产风险评估能力等;
9.涉及持有人转让的,转出方与受托方应当提供在规定时间内核减原批准委托生产范围的承诺书,以及相关情况说明。情况说明主要包括符合公告中第(十七)条的情况;
10.材料真实性声明;
11.申请企业对于业务办理人员的授权委托书。
附2:出具《同意受托生产意见书》的检查要求
一、受托生产线有已上市产品
受托生产生物制品、无菌药品等高风险药品,应当依据该品种或者所在生产线一年内检查结论为“符合要求”的药品GMP符合性检查结果告知书出具《同意受托生产意见书》;受托品种为其他类型药品的,应当依据该品种或者所在生产线五年内检查结论为“符合要求”的药品GMP符合性检查结果告知书,出具《同意受托生产意见书》。
二、受托生产线为新建生产线无已上市产品
可以依据一年内该生产线结论为“符合要求”的许可检查结果出具《药品受托生产意见书》。
上述“情形一”中告知书结果超出期限的应当组织开展药品GMP符合性检查,“情形二”中许可检查结果超出期限可组织药品GMP符合性检查或按许可标准开展检查,组织药品GMP符合性检查的可基于受托品种开展,符合要求后予以出具。新建、改建、扩建车间和生产线且已有上市产品的,可基于已上市品种开展药品GMP符合性检查。
受托生产线通过相应剂型GMP符合性检查,但没有下发药品GMP符合性检查结果告知书的,既往检查未发放GMP符合性检查告知书的,省级药品监督管理部门可结合既往检查结果办理《同意受托生产意见书》。如受托生物制品、无菌药品等高风险药品一年内通过相应剂型GMP符合性检查的报告。
附3:
附件2:意见反馈表